


Function prediction
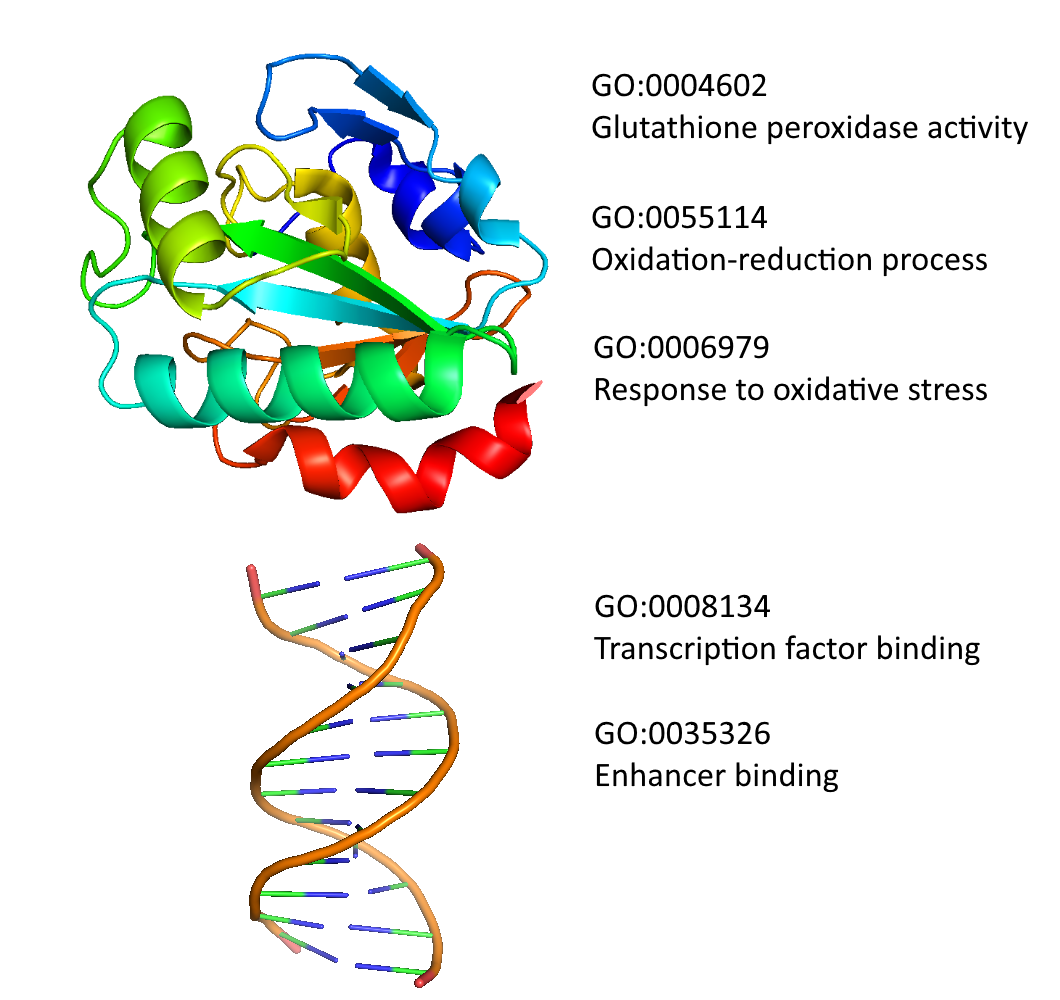
Our group has a long-lasting expertise in the development of methods for the prediction of gene and protein function. The first tool, Argot, dates back to 2009, and its further developments Argot2 and Argot2.5 successfully participated to the Critical Assessment of Protein Annotation (CAFA) in 2011 and 2014. The methods are available as freely usable web servers and accept submissions of thousands of sequences (see “Tools”). Currently, a new version of the algorithm is being implemented, in view of the third edition of the CAFA challenge that will start at the beginning of 2017.
Functional taxonomy
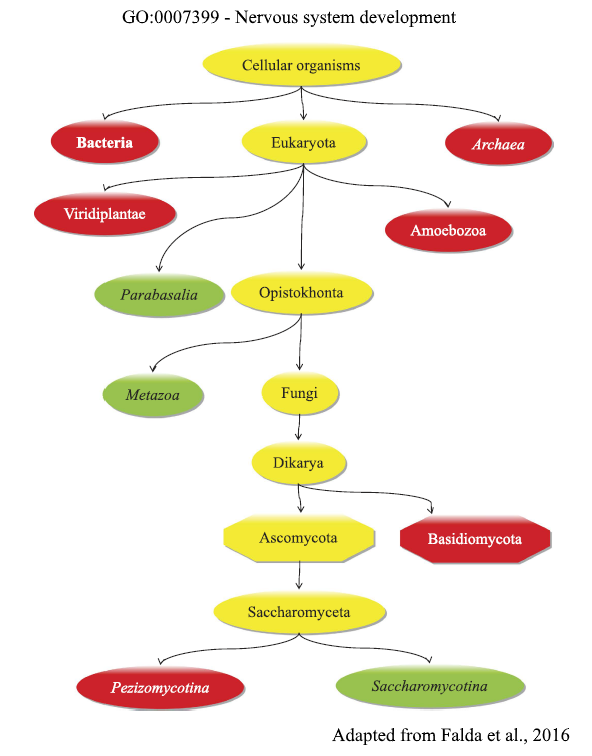
Started as a spin-off of the “Function prediction” project, this work aims to define the range of functions that a specific organism is able to perform by providing taxonomic constraints for Gene Ontology annotations. Starting from such information, it is possible to compare different species and evaluate specificities and commonalities existing among them. The final product, the Functional Taxonomy Information System (FunTaxIS), is available as web server and allows to explore the data at both GO-centric and a taxon-centric levels. Taxonomic constraints also help to refine function predictions, by preventing functional transfers of uncorrect annotations between distant organisms.
Whole genome sequencing and comparative genomics
 We participate to several whole genome sequencing projects performed with both Sanger and next-generation sequencing technologies. We collaborated with international consortia for the sequencing of apple (Malus domestica) and grapevine (Vitis vinifera) genomes, while more recently we are focusing on microbial and viral species for the identification of pathogenic islands, resistance genes, sequence variability among close strains, etc.
We participate to several whole genome sequencing projects performed with both Sanger and next-generation sequencing technologies. We collaborated with international consortia for the sequencing of apple (Malus domestica) and grapevine (Vitis vinifera) genomes, while more recently we are focusing on microbial and viral species for the identification of pathogenic islands, resistance genes, sequence variability among close strains, etc.
Viral genotyping as diagnostic tool
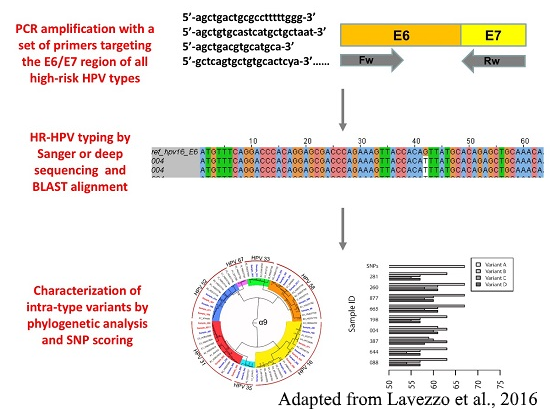
The genotyping of viral pathogens is important for several reasons, such as the management of patients, the evaluation of vaccines, the prognosis of some virus-related cancers, and for epidemiological surveys. Thanks to our close collaboration with the Virology and Microbiology units of our department, we carry on the development of methods for viral genotyping based on next-generation ultra-deep sequencing of PCR amplicons.
Viral epidemiology
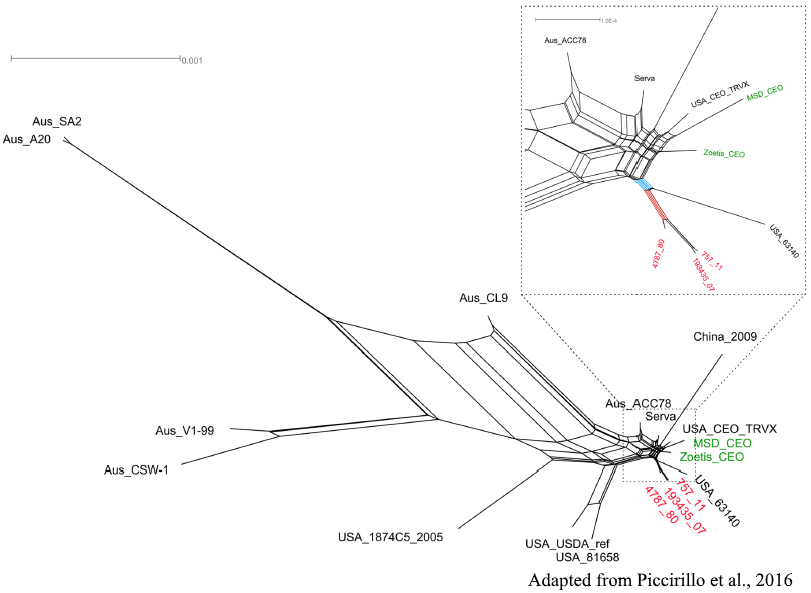
By applying whole genome or targeted sequencing, followed by phylogenetic and evolutionary analyses, we monitor the distribution and the variability over time of several viral pathogens, such as West Nile virus, Zika virus, Measles virus, Infectious laryngotracheitis virus, but also bacterial species like Mycobacterium tuberculosis.
Proteomics/Lipidomics
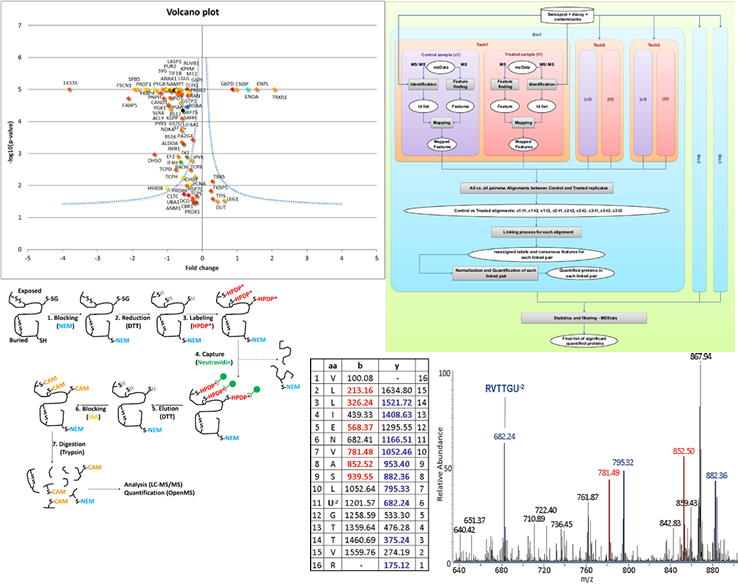
Our group is involved in the analysis and interpretation of Mass Spectrometry data in Proteomics. We are presently interested in the study of specific protein targets as Glutathione Peroxidases and the development of both new experimental approaches and data analysis pipelines for Redox Proteomics results. Recently, we are involved in the development of experimental techniques for the detection of lipids and their peroxidation products in the cell by using Mass Spectrometry and different tools for data analysis.
Biochemistry
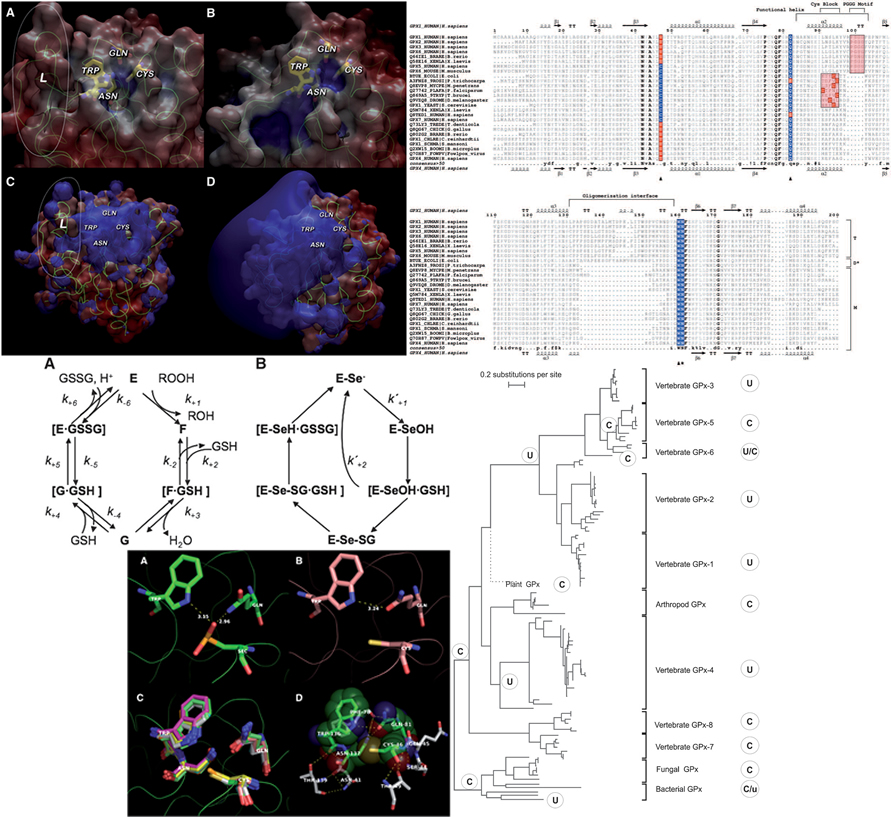
Our laboratory has a long-standing expertise in the study of protein families at in silico level as protein structure prediction, phylogenetic analysis, and catalytic mechanisms. We collaborate with different groups helping us to elucidate the inner working mechanisms of enzymes involved in the Redox regulation of the cell redox homeostasis. To this extent we have particularly focused our studies on Glutathione Peroxidase family members and the biological processes where they exert their functions interacting with other important players of redox regulation.
3D genome
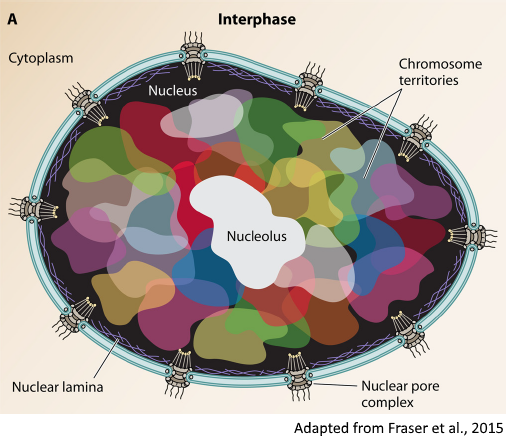
Recently, we approached a new line of research aimed at the characterization of the three-dimensional structure of genomes, which is a new attractive component of cell biology that is involved in many fundamental processes. Thanks to innovative experimental protocols (i.e. Hi-C), it is now possible to investigate the spatial conformation of chromosomes and identify interacting domains and long-range loops. The mechanisms underneath the formation of highly conserved chromosomal structures are still largely unknown, leaving many interesting biological questions to address.
RNA-seq and pathway analysis
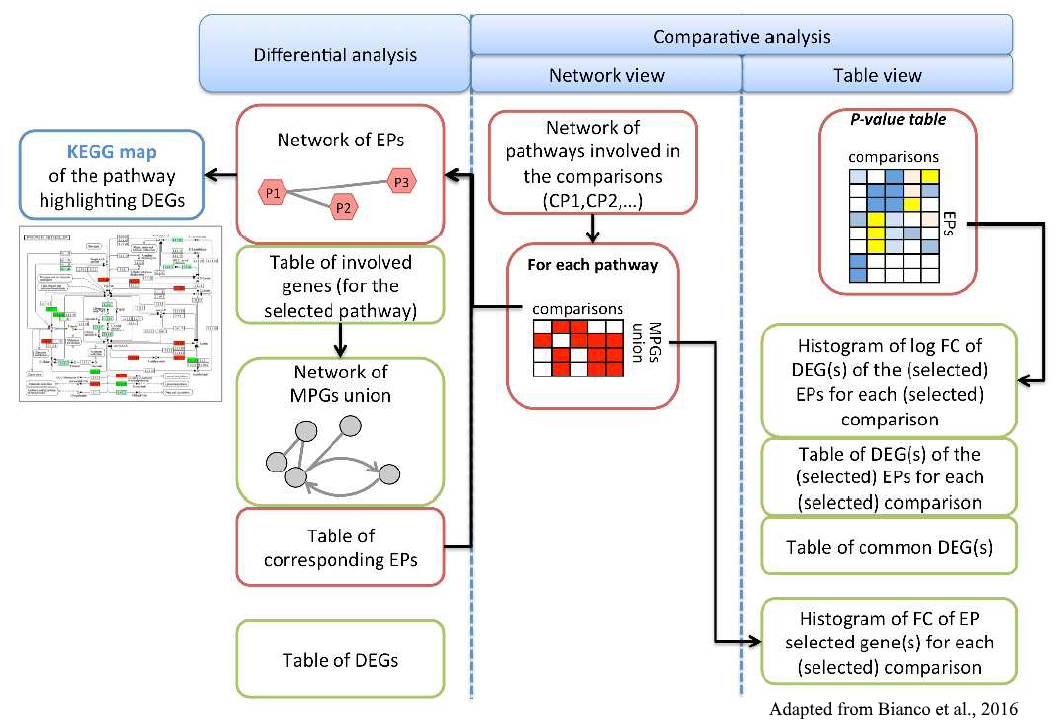 Additional projects, carried on in collaboration with other groups from inside and outside the University of Padova, relate to omics studies for the evaluation of gene expression patterns (RNA-seq) in different organisms (from plants to microbes). Distinct levels of functional characterization of the observed variability are being explored, from single genes to high-order metabolic and biological pathways. A web application is already available, called Pathway Inspector, designed to guide the user from raw expression data to pathways enrichment.
Additional projects, carried on in collaboration with other groups from inside and outside the University of Padova, relate to omics studies for the evaluation of gene expression patterns (RNA-seq) in different organisms (from plants to microbes). Distinct levels of functional characterization of the observed variability are being explored, from single genes to high-order metabolic and biological pathways. A web application is already available, called Pathway Inspector, designed to guide the user from raw expression data to pathways enrichment.